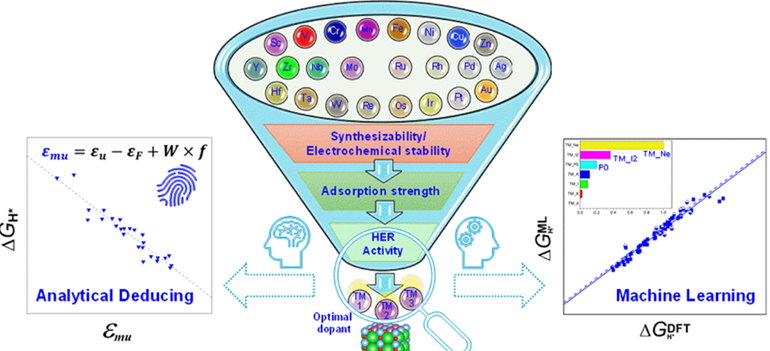
Doping regulation holds promise to modulate electrocatalytic performance, yet it remains largely unexplored for ferroelectric (FE) BaTiO3 (BTO). By jointly employing first-principles calculations and machine learning (ML) analysis, we examine the effect of a broad range of transition metal (TM) doping in FE BTO on the electrocatalytic hydrogen evolution reaction (HER) activity and screen out the optimal TM dopants. We unveil that some early-to-middle group TM (V, Cr, Mo, Ta, Ru)-doped BTO surfaces feature higher synthesizability, which also exhibit noticeable HER activity with |ΔGH*| < 0.2 eV owing to intermediate hydrogen adsorption strength. Among all doped surfaces, the Mo-doped one shows optimal HER activity under both out-of-plane and in-plane polarization states. We reveal an intense interplay between the hydrogen adsorption configuration and the corresponding hydrogen bonding interaction, which relies more on the TM group than the polarization state. Most importantly, we propose a physically informed descriptor of the surface oxygen p band, which better describes HER activity trends of TM-doped surfaces than conventional band descriptors.This indicates the significance of the fractional filling and bandwidth of occupied oxygen p-band states. Moreover, we establish a robust ML model that can well predict HER activity with surface-independent input parameters alone with R2 value above 0.93. From these parameters, we identify the inherent outer electron number of the TM dopant as the dominant feature, while the second ionization energy of the TM dopant and the initial polarization state show non-negligible feature importance. These findings could enlighten understanding, rational design, and accelerated discovery of element doping of FE materials for catalysis and other implications.